The ‘DNA Data Deluge’
Modern genomics has been a witness to an unprecedented data revolution in the last decade. Tens of thousands of sequencing machines worldwide sequence genomes at low cost, having generating exabyte-scale genome fragment data in remarkably short time. This development was coined the ‘DNA Data Deluge’ already in 2013. Still, this has just been the beginning. Significant sequencing technology shifts continue to routinely hit the market, every time skyrocketing sequence data volumes yet again.
Only now, it has become possible to analyze organisms at population scale, which finally sheds light on several topics of urgent interest. Also, the ultra-large data volumes bring about an overwhelming Increase in statistical power, important for genome-wide association studies (GWAS) and (previously inconceivable) machine learning strategies
Pan-Genomes
Individual genomes can be considered as strings over the alphabet {A,C,G,T}, which represent the nucleotides as fundamental building blocks of DNA. One realizes that significant parts of sequence are shared between pairs of individuals of a species, or, more generally, within evolutionarily coherent collections of genomes. For example, on average only 1 out of 1000 positions of an individual ~3.2 billion letter-long human genome varies in comparison with another individual.
A (biomedically/evolutionarily coherent) collection of genomes, large enough to capture all genetic variation of interest in the given context, is called a pan-genome. All humans of African origin, for example, give rise to the African pan-genome. Context specific and/or analysis specific pan-genomes are also conceivable, such as the pan-genome reflecting the microbiomes of, say, all people exposed to certain environmental factors, such as particular types of antibiotics.
The vast majority of the reference systems employed to date are linear sequences. But a linear reference genome cannot satisfyingly capture all types of variants. Examples are translocations—pieces of sequences that are transferred to other parts within genomes—since they break the linear order of genomes. Even for point mutations (single letter changes) and small insertion/deletion variants, only one of the possible options can be kept, which introduces statistically disturbing biases. More flexible, less biased approaches are imperative for a wealth of important applications.
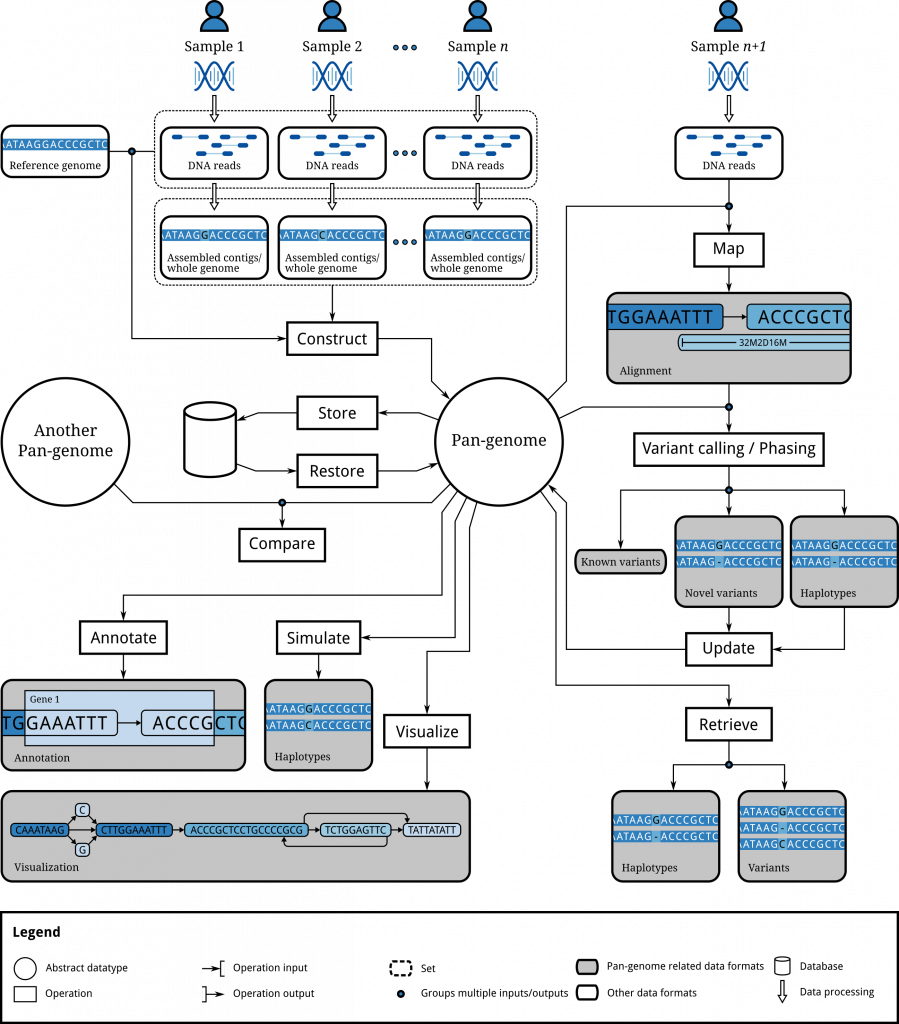
Solution: Computational Pan-Genomics (CPG)
Certain types of graphs offer promising solutions. The realization that new, graph-based types of reference systems need to be implemented has led to establishing the still emerging field of computational pan-genomics. Its goal is to offer computational solutions for pan-genome reference systems that support the duality of reference versus individual genome in a flexible and unbiased manner. Despite the urgency—exabytes of data keep being generated at accelerating pace—and despite the fact that pan-genomes are an established concept in biology, the computational analysis is still carried out mostly in an ad-hoc manner.
The paradigm shift from sequences to graphs requires to make substantial advances in terms of algorithms and data structures.
The figure to the right illustrates the transformation of a collection of related genomes (percentages reflect abundances within the population) into a ‘variation graph’, currently a predominant graph-based pan-genome representation. Notice that the variation graph is both smaller and more informative than the original set of plain linear sequences. However, the efficient construction of variant graphs and several other tasks (comparisons of variations graphs, detection of genetic variations within them, updates, etc.) are linked to difficult computational problems for many of which substantial improvements still can be expected.

The demand for novel, algorithmically fundamental is evident: usage of graph-based reference genome structures, although evidently beneficial in many ways, is largely uncommon and more complex. New efficient algorithms and data structures for building, managing, and comparing such genome graphs have to be designed. While software products for dealing with pan-genome data are needed right now to support clinicians and biologists, large-scale implementations are thwarted mostly due to the lack of algorithm development, insufficient software engineering efforts and unavailability of skilled researchers and staff.
Main research object
Leading the paradigm shift from sequence- to graph-based representations of genomes.
The ALPACA ITN raises a new class of researchers who master the complexity of the era of computational pan-genomics, and thus required to bring along an innovative, unique set of skills: being both highly interdisciplinary and multi-specialized, able to address problems ranging from fundamental algorithms and data structures to software development, big data management and analysis, statistics and machine learning, all closely entangled with genomics and genetics, and bioinformatics in general, and able to bring together the academic and industrial sectors engaged in related business.
We will provide new graph-based representations of evolutionarily related collections of genomes, together with the computational operations that implement their practical benefits. This is instrumental for leveraging the potential of the big genome data (and preventing serious congestion of resources). We will obtain decisive improvements in terms of
- Redundancy reduction and data compression
- The convenient highlighting of commonalities and differences
- Visualization
- Comprehensive annotation.
To amplify the benefits of those improvements we will provide software implementations of quality competitive with sequence-based software packages in terms of computational complexity. ALPACA will be focused on three intertwined directions:
- Constructing, updating, indexing and compressing pan-genome graphs (Work Package 1),
- Comparing pan-genome graphs, for instance by constructing and evaluating phylogenetic trees from single or multiple pan-genome graphs (Work Package 2),
- Transforming pan-genome graphs into input amenable to genome-wide association studies and machine learning based applications (Work Package 3).